

Summary
The current procedure thoroughly explains how to reprogram induced pluripotent stem cells (iPSCs) from the patient’s peripheral blood mononuclear cells (PBMCs), a less invasive source; e.g., somatic cells. We describe how to isolate PBMCs and reprogram them into iPSCs by electroporation. Furthermore, we provide an alternative approach to generating iPSC using Geltrex or Matrigel matrix to replace MEF-feeder. The challenge of this process is the relatively lower cell survival rates of PBMCs due to the damage of electroporation.
For complete details on the use and execution of this protocol, please refer to Hu et al. (2021).
Subject areas: Cell Biology, Health Sciences, Stem Cells
Graphical abstract
Highlights
-
•
Protocol to induce iPSC from patient-derived PBMCs
-
•
Protocol to isolate PBMC from patients
-
•
Alternative feeder approach to generating iPSC by Geltrex or Matrigel matrix
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
The current procedure thoroughly explains how to reprogram induced pluripotent stem cells (iPSCs) from the patient’s peripheral blood mononuclear cells (PBMCs), a less invasive source; e.g., somatic cells. We describe how to isolate PBMCs and reprogram them into iPSCs by electroporation. Furthermore, we provide an alternative approach to generating iPSC using Geltrex or Matrigel matrix to replace MEF-feeder. The challenge of this process is the relatively lower cell survival rates of PBMCs due to the damage of electroporation.
Before you begin
Detailed procedures for the generation of iPSCs from patient-derived PBMCs are described. Patients with the specific disease must first be chosen, in the case of the current protocol, PBMCs were harvested from patients with Dent I disease. The patient’s blood was collected following the criteria of the Declaration of Helsinki and experiments were authorized by the Medical Ethics Committees (2022-IRBAL-068). The use of pregnant mice was also authorized by Zhejiang University Experimental Animal Welfare Ethics Review Committee (22076). All procedures were carried out under sterile conditions in Class II biological hood. Mouse embryonic fibroblasts (MEFs), PBMCs, and iPSCs were all cultured in a humidified incubator at 37°C with 5% CO2.
The following procedures, consisting of three main steps before the generation of iPSCs, are time-consuming, labor-intensive, and expensive. Matrix-coated culture plates, medium, supplements, and solutions must all be prepared in advance. Dishes and plates for cell culture must be coated with extracellular matrix proteins or protein mixes for enhanced cell attachment. Antibiotics are not used in the iPSC growth media to avoid unknown outcomes (Choi et al., 2011; Hu et al., 2021; Loh et al., 2010; Mack et al., 2011; Niibe et al., 2011; Okita et al., 2013; Takahashi and Yamanaka, 2006; Yu et al., 2007; Zhang et al., 2001; Zhou et al., 2011).
Note: mTeSR™1 and Versene are used for iPSCs culture and passaging, respectively. Other commercial reagents such as E8 and ReleSR may be suitable alternatives. However, we have not conducted any tests, we could not guarantee that these alternative reagents would produce identical results.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-OCT3/4 (1:300) | Santa Cruz Biotechnology | Cat # sc-5279 RRID: AB_628051 |
| Mouse anti-NANOG (1:300) | Santa Cruz Biotechnology | Cat # sc-293121 RRID: AB_2665475 |
| Mouse anti-SOX2 (1:300) | Santa Cruz Biotechnology | Cat # sc-365823 RRID: AB_10842165 |
| PE anti-human TRA-1-81 (1:50) | BioLegend | Cat # 330708 RRID: AB_1089243 |
| APC anti-human SSEA-4 (1:50) | BioLegend | Cat # 330418 RRID: AB_2616819 |
| Donkey Anti-Mouse IgG H&L (Alexa Fluor 488 (1:400) | Abcam | Cat # ab150105 RRID: AB_ 2732856 |
| Biological samples | ||
| Patient blood sample | The Children’s Hospital, Zhejiang University School of Medicine | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| mTeSR™1 medium | STEMCELL Technologies | Cat # 85850 |
| Y-27632 RHO/ROCK pathway inhibitor | STEMCELL Technologies | Cat # 72304 |
| CHIR99021 | STEMCELL Technologies | Cat # 72052 |
| Human Recombinant Fibroblast growth factor 9, FGF9 | STEMCELL Technologies | Cat # 78161 |
| Heparin | STEMCELL Technologies | Cat # 07980 |
| Geltrex™, LDEV-Free | Gibco | Cat # A1413201 |
| Corning Matrigel hESC-qualified Matrix | Corning | Cat # 354277 |
| CryoStor CS10 | STEMCELL Technologies | Cat # 07959 |
| StemSpan™-XF | STEMCELL Technologies | Cat # 100-0073 |
| StemSpan™ CC100 | STEMCELL Technologies | Cat # 02690 |
| DMEM-high glucose | Sigma | Cat # RNBK3725 |
| Mitomycin C | STEMCELL Technologies | Cat # 73272 |
| Versene | Gibco | Cat # 15040-66 |
| TrypLE select | Gibco | Cat # 12563029 |
| Gelatin | YEASEN | Cat # 40108ES60 |
| Critical commercial assays | ||
| P3 Primary Cell 4D-Nucleofector™ X Kit | Amaxa Lonza | Cat# V4XP-3012 |
| Amaxa Human T Cell Nucleofector Kit | Amaxa Lonza | Cat # VPA-1002 |
| PBMC Isolation Kit | Solarbio | Cat # P8610 |
| DNA Isolation Mini Kit | Vazyme | Cat # DC102-01 |
| PCR Mycoplasma Detection Kit | TransDetect® | Cat # FM311-01 |
| Experimental models: Organisms/strains | ||
| Mouse: CF1 13.5 days pregnant | Hangzhou, Xuanzhu Bio | CF1 |
| NOD/SCID Immune-deficient mice | Hangzhou, Xuanzhu Bio | N/A |
| Oligonucleotides | ||
| Primer for episomal plasmid Orip F: TTCCACGAGGGTAGTGAACC R: TCGGGGGTGTTAGAGACAAC (544 bp) |
Hangzhou, Youkang Bio | N/A |
| Primer for pluripotency EBNA1 F: ATCAGGGCCAAGACATAGAGATG R: GCCAATGCAACTTGGACGTT (96 bp) |
Hangzhou, Youkang Bio | N/A |
| Other | ||
| Fluorescent microscopy | ZEISS | 880 |
| CytoFLEX LX Flow Cytometer | Beckman Coulter | |
| 4D-Nucleofector | Lonza | |
| Coverslips | Solarbio | Cat # YA0350 |
Materials and equipment
PBMC culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | – | 44.5 mL |
| FBS | 10% | 5 mL |
| Penicillin-Streptomycin | 1% | 500 μL |
| Total | – | 50 mL |
PBMC cryopreservation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| FBS | 90% | 9 mL |
| DMSO | 10% | 1 mL |
| Total | – | 10 mL |
MEF culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM-high glucose | – | 44.5 mL |
| FBS | 10% | 5 mL |
| Penicillin-Streptomycin | 1% | 500 μL |
| Total | – | 50 mL |
Reprogramming medium
| Reagent | Final concentration | Amount |
|---|---|---|
| StemSpan™-XF | – | 6.3 mL |
| StemSpan™-CC100 | 10% | 700 μL |
| Total | – | 7 mL |
Note: Thaw StemSpan™ CC100 and StemSpan™-XF at 4°C, mix thoroughly.
Reprogramming plasmids
| Reagent | Final concentration | Amount |
|---|---|---|
| HoCT3/4 | 1000 ng/μL | 1 μg |
| Hsk sox2 KLF4 | 1000 ng/μL | 1 μg |
| L-myc LIN28 | 1000 ng/μL | 1 μg |
| Total | – | 3 μg |
4D-Nucleofector™ X Unit (V4XP-3012)
| 100 μL single nucleocuvette | Final concentration |
|---|---|
| PBMCs | 1–5∗10ˆ6 |
| plasmid DNA | 1–5 μg |
| P3 Primary Cell 4D-Nucleofector™ X Solution | 100 μL |
| Program | EO-015 |
Geltrex™ matrix
-
•
Add pre-chilled (4°C) Geltrex™ Matrix solution to DMEM/F-12 medium following the optimal coating concentration for your application empirically (1:50–1:200).
-
•
Cover the surface of the dish with sufficient Geltrex™ Matrix solution. For example, add 1.5 mL for a 35 mm dish, or 3 mL for a 60 mm dish.
-
•
Before use, incubate the coated-plates at room temperature (RT, 15°C–25°C) for about 30–60 min.
-
•
Carefully remove the supernatant, then immediately place cells in a pre-equilibrated cell culture medium.
Note: We have experimented with coating concentrations ranging from 1:50 to 1:200. In our application, 1:100 is recommended for iPSCs propagation, while 1:50 is recommended for iPSCs reprogramming. Thaw Geltrex™ Matrix at 2°C–8°C overnight, then aliquot and store at −20°C.
CRITICAL: To minimize premature gelling, wrap the coated dish in Parafilm sealing film and keep it at 4°C for two weeks after step 2. Next time before use also needs to start from step 3. Do not allow the coated surface to dry out.
Corning Matrigel hESC-qualified matrix
-
•
Add one aliquot of Matrigel matrix to 25 mL of DMEM/F-12.
-
•
Incubate the plates at RT for 30 min to 1 h before use.
-
•
Aspirate the remaining liquid from dishes just before use. Ensure that the tip of the pipet does not scratch the coated surface.
-
•
The coated dish is stable within two weeks when wrapped with Parafilm™ sealing film and stored at 4°C.
Note: Thaw Corning Matrigel hESC-qualified Matrix at 4°C overnight. Once Matrigel Matrix is thawed, gently swirl the vial to ensure that Matrix is evenly dispersed. Based on the protein concentration from different batches, the volume of the aliquots varies from 270–350 μL.
mTeSR™1 medium for iPSC
Add 100 mL 5× Supplement to 400 mL mTeSR™1 Basal Medium and mix thoroughly.
Note: If not used immediately, store the complete mTeSR™1 medium at 2°C–8°C for up to 2 weeks. Alternatively, store at −20°C for up to 6 months.
ROH/ROCH pathway inhibitor Y-27632 (10 mM)
-
•
Dissolve 1 mg Y-27632 in 624 μL D-PBS (pH 7.2).
-
•
To avoid several freeze-thaw cycles, aliquot into working volumes (20 μL each).
-
•
The stock solutions should be diluted with culture medium at a final concentration of 10 μM before use.
Gelatin (0.1%)
-
•
Take 2 g of gelatin powder into 200 mL of PBS.
-
•
Heat with water bath until fully dissolved (42°C–65°C).
-
•
Put the prepared gelatin solution (1%–2%) into the glass bottle and autoclave for 121°C, 20 min, at the pressure of 15 psi. After sterilization, it could also be stored at 4°C for 4–8 weeks.
-
•
Dilute into 0.1% with ddH2O or PBS before use. Do not warm it before using it.
-
•
Put 1 mL 0.1% gelatin solution into the 6-well plate, and incubate at RT for 30–60 min.
-
•
Before use, remove the gelatin solution and wash twice with D-PBS.
-
•
The gelatin coated-plates can be stored at 4°C for 4–8 weeks.
Mitomycin C (10 μg/mL)
-
•
Prepare stock solutions with PBS before use.
-
•
Dilute 5 mg mitomycin C with 50 mL PBS (100 μg/mL) and store at −20°C (10 μL) aliquots.
-
•
For use as a cell culture supplement, stock solutions should be diluted to 10 μg/mL with MEF culture medium (10% FBS and 1% Penicillin-Streptomycin-DMEM) before use (see “materials and equipment” for more information).
Step-by-step method details
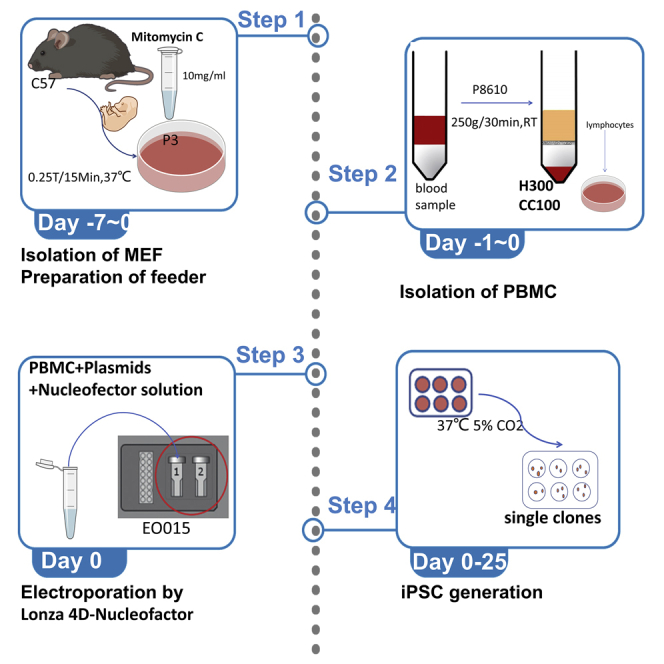
We have outlined a timeline scheme during the process of reprogramming iPSCs for better comprehension for beginners (Figure 1). Specific details are discussed below.
Figure 1.

The timeline scheme with the landmarks during the reprogramming process
Isolation of PBMCs
Timing: 2–4 h, day -1
Note: Isolate PBMCs by gradient centrifugation via PBMC Isolation Kit (see key resources table).
-
1.
Collect 2–4 mL of human venous blood in heparinized vials.
-
2.
The sample is diluted with an equal volume of D-PBS and transferred to a new 15 mL tube by gently mixing 2–5 times by inversion.
-
3.PBMCs are isolated according to the Solarbio P8610 technique.
-
a.Add an appropriate amount of separation solution into the centrifuge tube.Note: if the diluted blood volume (with PBS after step 2) is less than 3 mL, add 3 mL of separation solution; if the diluted blood volume is greater than or equal to 3 mL, add an equivalent volume of separation solution. However, the total volume should not exceed two-thirds of the 15 mL tube (10 mL), otherwise, the effect of centrifugation would be compromised.
-
b.Slightly pave the diluted blood above the solution’s surface.
-
c.Centrifuge at 500–1000 g for 20–30 min at RT with the horizontal rotor.Note: The larger the volume of blood, the higher the centrifugal force required to achieve a better outcome. The best separation conditions need to be explored and the maximum centrifugation speed should not be more than 1200 g. For example, for 8 mL total volume (the diluted blood and separation solution), we use 500 g, 30 min.CRITICAL: The amount of separation solution should be equal with the diluted blood sample (with D-PBS). If the total volume (the diluted blood sample and separation solution) is more than 10 mL, the diluted blood sample (with PBS) should be split into two tubes with an equal separation solution.
-
d.After centrifugation, from top to bottom, there are the diluted plasma layer, transparent separator layer, lymphocyte layer, and red blood cells and granulocytes (Figure 2). Gently absorb the lymphocyte layer by a plastic Pasteur pipette (3 mL Pasteur pipette) into a new 15 mL tube.
-
e.Wash the lymphocyte layer with 10 mL D-PBS by pipetting the cells up and down 8–10 times (3 mL plastic Pasteur pipette is recommended).
-
f.Centrifuge at 250 g for 10 min at RT.
-
g.Remove supernatant and resuspend the cell pellet in 10 mL D-PBS. Centrifuge at 250 g for 10 min at RT. Repeat this step.
-
h.Discard the supernatant and resuspend the cell pellet with 500 μL D-PBS. Count the number of cells using a hemocytometer.
-
a.
-
4.
PBMCs are frozen in cryopreservation solution or prepared at a density of 1–5 ∗10ˆ6 cells/mL for electroporation.
Note: Thaw PBMCs before electroporation and cultured with a PBMCs culture medium at least one night. After electroporation the mixtures including PBMCs and plasmids should be seeded into a 6-well plate coated with 1% (v/v) Matrigel, Geltrex matrix, or MEF-feeder.
Figure 2.

A cartoon indicating the four layers of blood after centrifugation with separation solution
(A) The top layer (yellow) is the diluted plasma layer.
(B) The middle (white) is the separation solution layer.
(C) The white membrane layer between the plasma and the isolate is the lymphocyte layer.
(D) At the bottom of the centrifugal tube is the red blood cells and granulocyte.
PBMCs electroporation and generation of iPSCs
Note: After many attempts, we found that Geltrex or Matrigel matrix may be a good substitute for MEF-feeder. Whereas the MEF-feeder may be more economical, Geltrex or Matrigel have defined compositions that reduce unknown effects. The steps 24–41 involving the isolation of MEF and preparation of the MEF-feeder are not essential according to the use of MEF-feeder or feeder-free culture. We recommend the use of the feeder-free culture on the grounds of simplicity and ease of use.
-
5.Electroporation of PBMCs may be carried out by Lonza 4D-Nucleofector™ using program EO-015, following the manufacturer’s protocol. (see “materials and equipment” for more information).
-
a.Prepare 6-well plates with 1 mL Geltrex/DMEM-F12 or 2∗10ˆ4 cells/cm2 MEF-feeder (see steps 24–41).
-
b.Pre-incubate desired volume (maximum 2 mL) of reprogramming medium (StemSpan™-XF with 10% StemSpan™-CC100) at a 37°C water bath.
-
c.Prepare the DNA plasmid solution with a concentration of 1 μg/μL in ddH2O. The plasmids containing hoCT3/4, hsk sox2 KLF4, and L-myc LIN28 encode the reprogramming factors.
-
d.Centrifuge the required number of PBMCs (1–5∗10ˆ6 cells) at 200 g for 10 min at RT. Remove supernatant completely.
-
e.Carefully resuspend the cell pellet at RT with 100 μL 4D Nucleofector™ Solution containing the recommended amounts of DNA plasmid (1 μg of each reprogramming plasmid, 3 μg of total DNA).
-
f.Transfer mixtures containing 1–5∗10ˆ6 number of PBMCs, 100 μL transfection solution, and required DNA plasmid into the Nucleocuvette™ Vessels. Start the program EO-015.
-
a.
-
6.
After the program has finished (about 3–5 s), take the Nucleocuvette™ Vessel out of the retainer and immediately place it in a humidified incubator for 5 min.
-
7.
Gently pipette up and down 2–3 times to mix cells. When working with the 100 μL Nucleocuvette™, use the pipettes supplied and avoid repeated aspiration of the sample.
-
8.
Seed the transfection cell suspensions into a 6-well plate coated with Geltrex or MEF-feeder. Culture the cells in reprogramming medium (See table "Reprogramming medium" of “materials and equipment” for more information).
-
9.
After 4 h of incubation, replace the medium with fresh reprogramming medium. Collect the cells and medium from each well into a 15 mL tube and centrifuge at 300 g for 10 min. Remove the supernatant and resuspend the cell pellet with 2 mL of fresh reprogramming medium. This step is necessary to ensure the removal of transfection solution which may damage the cells. This timepoint is defined as Day 0.
-
10.On day 3, replate cells into a new Geltrex or MEF-feeder coated 6-well plate.
-
a.Harvest the cell suspension into a tube and wash each well with 2 mL of D-PBS, aspirate it and add 700–800 μL TrypLE select reagent to detach the MEF-feeder cells and attached PBMCs. Incubate them for 4 min at 37°C.
-
b.Transfer dissociated cells into previously harvested cell suspension. Centrifuge at 300 g, 10 min, at RT.
-
c.Remove the suspension and resuspend the cell pellets with 2 mL of fresh reprogramming medium before transferring them to a new 6-well plate coated with fresh Geltrex or MEF-feeder.
-
a.
-
11.
On day 4, add 200 μL mTeSR™1 per well.
-
12.
On days 6, 8, and 10, collect 1 mL of medium and centrifuge at 300 g for 5 min at RT. Remove the supernatant, resuspend the cell pellets in 1.2 mL of mTeSR™1 and replate cells into the original well.
-
13.
On day 11, the medium is replaced completely with mTeSR™1. The culture medium should be replaced daily by simply aspirating the media and adding 2 mL fresh mTeSR™1.
-
14.
Day 12–25: Perform daily medium changes (2 mL/well) using mTeSR™1. Monitor the cells until iPSC colonies appear.
Note: iPSC colonies typically form 10–20 days after electroporation. At 4× magnification, the cells appear as packed clusters under the microscope. For a representative example of an iPSC colony (Figure 3).
-
15.
iPSC clones should be identified and selected for amplification (step 16) when colony boundaries become blurred (Figure 3).
Figure 3.

The milestones of PBMC reprogramming into iPSC
The DAY 9\10\11\16\18\20 after electroporation, iPSC clones should be identified and selected for amplification when their boundaries become blurred show in DAY 20. Magnification 4×, scale bar 50 μm.
Selection and amplification of iPSCs
Note: Prewarm mTeSR™1 medium and 96-well plates coated with Matrigel or Geltrex at RT (15°C–25°C) before operation.
-
16.
Under a microscope at 10× magnification, remove the majority of the medium, leaving about 100–200 μL.
-
17.
Break up the putative iPSC colony into small fragments using a pipette. Scrape and aspirate the colony fragments using a 200 μL pipettor.
-
18.
Seed the iPSC colony fragments into a 96-well plate coated with Geltrex or Matrigel matrix. Culture with 100 μL mTeSR™1 and 10 μM Y-27632 at 37°C in a 5% CO2 incubator.
-
19.
After 24 h, replace the medium with 100 μL mTeSR™1 without Y-27632.
-
20.
The culture medium should be changed daily with 100 μL fresh mTeSR™1.
-
21.
Cells should be passaged when they achieve 70% confluence.
-
22.
Gradually passage the iPSCs into 48\24\12\6-well plates. For example, after 2–3 passages in 96-well plates, iPSCs should be amplified into 48-well plates with the ratio of 1:1.
-
23.
When cell numbers are enough (at least six 6-well plates), we prefer performing functional verification tests.
Isolation of mouse embryonic fibroblast (MEF)
Note: The steps 24–41 involving the isolation of MEF and preparation of the MEF-feeder are optional and are not needed when coated plates are chosen instead.
Harvest embryos at embryonic day (E) 13.5 from CF-1 mice.
Note: Prepare 0.1% gelatin to coat-plates (5 mL 0.1% gelatin per 10 cm dish) and incubate in 37°C for 30 min (see materials and equipment).
-
24.
Pregnant mice (day 13.5) are sacrificed by cervical dislocation and immersed in 75% ethanol.
-
25.
Cut through the full thickness of the skin with sterile scissors, then pull back the skin to expose the abdominal wall.
-
26.
With fresh sterile scissors, cut through the abdominal wall. Use forceps to lift the uterine horns and excise the uterus with scissors. Avoid allowing the uterus to come into contact with the fur. Place the uterus in a 50 mL tube with 30 mL DMEM with antibiotics to avoid potential contaminants from the mice.
-
27.
Separate the day 13.5 embryos and transfer them to a new 10 cm dish with fresh DMEM or D-PBS to wash out the blood.
-
28.
Discard the head, heart, liver, and four limbs. Mince the remaining embryos as finely as possible (about 1 mm3 pieces) with a scalpel and digest in 50 mL tubes with 0.25% trypsin-EDTA (use 10 mL 0.25% trypsin EDTA for every 2–3 embryos).
-
29.
Put the tube in a 37°C water bath for 15–20 min, mixing the cells every 3–5 min.
-
30.
Put the cell suspension and equal volume of medium (DMEM+10% FBS) onto a plate to inactive the trypsin. Homogenize by pipetting with a 1 mL pipet 8–10 times until no lumps are visible.
-
31.
Filter the cell suspension through a 70 μm strainer into a new tube.
-
32.
Centrifuge at 300 g, 5 min at RT. Discard the supernatant and re-suspend the cell pellet in a 1 mL MEF culture medium. Transfer the cell suspension to 10 cm dishes pre-coated with gelatin and add 9 mL medium.
-
33.
Incubate cells at 37°C in a humidified 5% CO2 incubator.
-
34.
Refresh medium daily and passage every 1–3 days when the cells reach 70% confluence (Figure 5). The passage ratio should be 1:4 to 1:7 (appropriately 2∗10ˆ4 cells/cm2).
-
35.
After 2 or 3 passages, non-adherent cells may be removed by aspirating the medium and the resulting MEFs may be utilized as feeder cells after mitotic inactivation using mitomycin C.
Note: The protocol of passage, cryopreservation, and thawing of MEFs are described in the final parts of this protocol.
Figure 5.

MEF cells cultured in 0.1% gelatin-coated plates
(A) MEF reaches 80%–90% confluence at the optimal time of passage.
(B) MEF after passage. Magnification 4×, scale bar 50 μm.
Preparation of the feeder
Note: Prepare 0.1% gelatin to coat-plates (10 cm dish) and incubate in 37°C incubator for 30 min.
-
36.
After the growth of the 3rd passage MEFs to 80%–90% confluence, remove the medium from the 10 cm dish and rinse cells twice with 10 mL of D-PBS.
-
37.
Add 10 mL of freshly prepared MEF inactivation medium, consisting of 9 mL of MEF medium and 1 mL 100 μg/mL mitomycin C (final concentration 10 μg/mL mitomycin C). Incubate cells for 2–4 h in a 37°C incubator.
-
38.
Remove the medium containing mitomycin C and rinse 3–5 times with 8–10 mL of D-PBS.
-
39.
Detach MEFs with 3 mL 0.25% trypsin-EDTA and incubate at 37°C for 1 min. Add 3 mL medium to inactivate the trypsin-EDTA and transfer to a 15 mL tube. Centrifuge at 300 g for 5 min at RT, and discard the supernatant.
-
40.
Resuspend cells with MEF culture medium and adjust cell density.
-
41.
Add cell suspension to a 6-well plate precoated with 0.1% gelatin (about 2∗10ˆ4 cells/cm2) and culture in a humidified 5% CO2 incubator.
Note: As indicated in steps 24–41, prepare the MEF-feeder seeded in 6-well plates with 2∗10ˆ4 cells/cm2 and culture with MEF culture medium until PBMCs are fed in. (See “materials and equipment” for more information.).
Analysis of iPSC
Immunofluorescence staining
-
42.
Place glass coverslips in a 24-well (14 mm) plate and coat them with Geltrex or Matrigel as described in the materials and equipment.
-
43.
Split iPSCs as detailed in supplement protocol “iPSC passaging” and plate them into prepared coverslips.
Note: The diluted ratio can range from 1:60 to 1:100, for example, iPSC reaches 80%–90% confluence of a well of 6-well plate can be diluted into 1:80 and plate into 14 mm coverslips.
-
44.
1–3 days later (when iPSC colonies acquire round shape morphology with smooth edges), remove supernatant and wash cells with PBS twice. (Figure 6).
-
45.
Fix cells with 4% (w/v) paraformaldehyde (PFA) for 20 min at RT.
-
46.
Wash cells with PBS twice.
-
47.
Permeabilize the samples by incubating coverslips for 10 min with PBST-(PBS containing 0.3% Triton X-100) at RT. Wash with PBS three times.
-
48.
Block cells with PBS containing 4% bovine serum albumin at RT for 30–60 min.
-
49.
Use the blocking media mentioned above (PBS containing 4% bovine serum albumin) to dilute the primary antibody (the ratio of dilution is according to the manual of antibody and it is detailed in the reagents table).
-
50.
Incubate the cells with diluted primary antibody (anti-OCT3/4, anti-NANOG, anti-SOX2) at 4°C overnight.
-
51.
Wash the cells with PBS for 10 min three times.
-
52.
Incubate samples with secondary antibodies (Donkey Anti-Mouse IgG H&L Alexa Fluor 488) diluted into blocking media for 1 h at RT in the dark.
-
53.
Coverslips were mounted onto glass slide with Dapi-Fluoromount-G (Thermo Fisher Scientific). Stained cells can be observed via a Carl Zeiss LSM 880 confocal microscope (Figure 7).
Figure 6.

1–3 days after iPSC passage into coverslips when iPSC colonies acquire round shape morphology with smooth edges
Figure 7.

Representative images show the pluripotency of iPSC indicated by OCT3/4, NANOG, and SOX2
Flow cytometry analysis
-
54.
iPSCs grown in a 6-well plate may be harvested with 0.5 mM EDTA (Versene), as described in the supplement protocol “passaging iPSC”.
-
55.
Wash cell pellets twice in cold PBS and centrifuge at 300 g for 5 min at RT.
-
56.
Remove the supernatant, resuspend into single cells with 100 μL PBS, and incubate the single cell with 100 μL 2% FBS-PBS for 10 min at RT.
-
57.
Centrifuge at 300 g, 5 min at RT. Remove the suspension and incubate the cells with the primary antibody (PE anti-human TRA-1-81, APC anti-human SSEA-4) for 30 min on ice.
-
58.
After staining, wash cells twice with PBS and analyze by CytoFLEX LX Flow Cytometer (Beckman Coulter).
Teratoma formation
Use cell detachment solution (Versene) to harvest iPSCs (typically obtained from a 70% confluent of 6-well plate) and resuspend cells in Matrigel diluted (1:1) with 200 μL DMEM/F12.
-
59.
Inject cells subcutaneously into the rear haunch of NOD/SCID immunodeficient mice using a 1 mL syringe.
-
60.
At 8–12 weeks after iPSC injection, dissect and fix teratomas in 10% formalin.
-
61.
Microsection samples and stains with hematoxylin and eosin (HE) for analysis.
Note: Please see our previous article (Hu et al., 2021) for more validated methods, including Karyotyping, Sanger sequencing, Short tandem repeat (STR) analysis, and Mycoplasma detection.
Supplement protocol of culture MEF and iPSC
Thawing MEFs
-
62.
Take out the cells from the liquid nitrogen and quickly thaw cells in a 37°C water bath by gently shaking the vial.
-
63.
Transfer the thawing cell culture medium to a 15 mL tube prepared with a 3 mL MEF culture medium. Gently mix the cell suspension with the medium with a 1 mL micropipette.
-
64.
Centrifuge at 300 g for 5 min at RT.
-
65.
Aspirate the medium, leaving the cell pellet intact. Gently resuspend the cell pellet in a 1 mL MEF culture medium with a 1 mL micropipette and place it into a gelatin-coated dish (about 2–4 ∗10ˆ4 cells/cm2). Add enough medium to the chosen plate and culture in a 37°C incubator.
Passaging MEFs
-
66.
Remove the medium and wash MEF with D-PBS 1–2 times.
-
67.
Add enough 0.25% trypsin-EDTA to the dish (3 mL for a 10 cm dish). Incubate for 1–2 min at 37°C.
-
68.
When cells start becoming round and detached, add an equal volume of MEF culture medium to the plate (see materials and equipment) to inactive the trypsin and pipet up and down with a 10 mL serological pipet 8–10 times and transfer cell suspension to a 15 mL tube.
-
69.
Centrifuge at 300 g for 5 min at RT. Discard the medium and resuspend the cell pellet in a 1 mL MEF culture medium.
-
70.
Transfer the cell suspension (appropriate 2∗10ˆ4 cells/cm2) to 10 cm dishes and add enough medium to each dish (8–10 mL).
Cryopreserving MEFs
-
71.
When MEFs grow to 80%–90% confluence or MEF-feeder is prepared, they can be frozen.
-
72.
Remove the medium of the dish and wash cells with D-PBS twice. Add required 0.25% trypsin-EDTA (1 mL /6-well plate, 3 mL/10 cm dish), incubate at 37°C for 1–2 min.
-
73.
Add an equal volume of MEF culture medium to inactivate the trypsin and collect the cells into a new 15 mL tube.
-
74.
Centrifuge at 300 g, 5 min at RT.
-
75.
Discard the supernatant, resuspend the cell pellet with MEF cryopreservation solution (see materials and equipment) (1 mL for a 10 cm dish, 300–500 μL for a well of 6-well plate).
-
76.
Put the cells suspension into cryogenic Vials (1∗10ˆ6 cells/vial) and a freezing container for freezing cells at a controlling rate.
Thawing iPSC
-
77.
Take out the cells from the liquid nitrogen and quickly thaw cells in a 37°C water bath by gently shaking the vial.
-
78.
Transfer the thawing cell culture medium to a 15 mL tube prepared with a 3 mL mTeSR™1. Gently mix the medium.
-
79.
Centrifuge at 300 g for 5 min at RT.
-
80.
Aspirate the medium, and gently resuspend the cell pellet in 1 mL of mTeSR™1 with 10 μM Y-27632 using a 2 mL pipette. Transfer cell suspension into a Geltrex or Matrigel-coated plate (about 1–5∗10ˆ4 cells/cm2). Add enough medium and culture in a 37°C incubator. Do not disturb the plate for 24 h.
Passaging iPSC
-
81.
Remove the medium and wash iPSC with D-PBS 1–2 times.
-
82.
Add enough Versene to the dish (1 mL for a 6-well plate). Incubate for 1 min at RT. Remove the Versene and incubate the cells at 37°C incubator for 4 min.
-
83.
When the cell mass begin to dissociate and detach, add 3–4 mL mTeSR™1 with 10 μM Y-27632.
-
84.
Carefully pipette the cell mass mixture up and down 2–3 times using a 2 mL pipette.
-
85.
Plate the cell suspension at the desired density onto coated wells containing mTeSR™1 with 10 μM Y-27632. The passaging ratio range from 1:6 to 1:20 and the mean aggregate size is approximately 50–200 μm).
-
86.
Place the plate in a 37°C incubator. 24 h later, change the medium into mTeSR™1 without 10 μM Y-27632.
Cryopreserving iPSC
-
87.
When iPSC grows to 80%–90% confluence. Remove the medium and wash iPSC with D-PBS 1–2 times.
-
88.
Add enough Versene to the dish (1 mL for a 6-well plate). Incubate for 1 min at RT. Remove the Versene and incubate the cells at 37°C incubator for 4 min.
-
89.
When the colony edges begin to loosen and lift from the dish, add 1 mL mTeSR™1.
-
90.
Gently add 2–3 mL DMEM/F12 to wash cells, and collect them into a fresh 15 mL tube.
-
91.
Centrifuge at 300 g, 5 min at RT.
-
92.
Discard the supernatant, and resuspend the cells with iPSC cryopreservation solution (CS 10) (1 mL for a 10 cm dish, 300–500 μL for a well of 6-well plate).
-
93.
Put the cells suspension into cryogenic vials and freezing containers for freezing cells at a controlling rate. Cells on each well of the 6-well plate can be frozen into 2–3 vitals.
-
94.
Next day, put the cryogenic vials into liquid nitrogen for long-term storage.
Expected outcomes
This protocol allows the generation of iPSCs from patient-derived PBMCs. The establishment of an iPSCs line will undoubtedly provide researchers and clinicians to build disease modeling. According to the current step-by-step protocol, iPSCs colonies appear 15–25 days after transfection by electroporation (Figure 3). iPSCs are culture in mTesR™1 and passage with Versene (Figure 4).
Figure 4.

iPSC passage by Versene
(A) The density of iPSC is 70%–90% confluence before passage.
(B) iPSC incubated in Versene after 5 min. Magnification 4×, scale bar 50 μm.
Limitations
Although electroporation gives a better transfection efficiency than other methods, the cell survival rate is much lower as a result of the damage caused by electroporation. Furthermore, specialist equipment is required for this technique which may not be available in each lab.
Troubleshooting
Problem 1
The number of MEF cells extracted from CF-1 is very low.
Potential solution
This may be due to an error in calculating the days of pregnancy, resulting in embryos that are too small for cell harvesting. However, the number of days of pregnancy should not be extended beyond 16 days.
Problem 2
The electroporation efficiency is low after the thawing of PBMCs.
Potential solution
Cryopreservation of PBMCs can damage cell viability. Performance of electroporation transfection immediately after the isolation of PBMCs or the day following overnight culture is recommended to greatly improve iPSC reprogramming efficiency.
Problem 3
iPSCs are easily contaminated.
Potential solution
iPSCs are cultured without antibiotics to avoid unknown effects which lead to easily be contaminated. The incubator and all consumables and reagents should be sterile.
Problem 4
Too many differentiated cells can be seen around the iPSC colonies under the microscope (5%–10% of the total area occupied by cells, Figure 8). Spontaneous differentiation is characterized by loss of colony border integrity, regions of irregular cell morphology within a colony, and/or the emergence of other identifiable cell types.
Figure 8.

Differentiated cells around iPSCs colonies
The emergence of cell types with different morphologies in the edge. The space of the clones in the center becomes loose.
Potential solution
If colonies are passaged too early or too frequently, cell Mass may not attach satisfactorily when replated, causing cells to start to differentiate (cell types with different morphologies emerge). If colonies are passaged too late, the culture may also begin to show signs of differentiation.
-
•
An approximate 24 to 48-h window is optimal for passaging. If there are large colonies with dense centers, the passage of the cells within 24 h (see Figure 4A for a representative image).
-
•
Ensure cells within areas of differentiation are removed before passaging. They can be removed with a pipettor under a stereoscope. (Under a microscope at 10× magnification, remove the majority of the medium, leaving about 100–200 μL. Break up the differentiated areas into small fragments using a pipette. Scrape and aspirate the fragments using a 200 μL pipettor.).
-
•
Ensure cultures are passaged when the majority of colonies are large, compact, and have dense centers relative to the edges. Do not allow the cells to become overgrown (Figure 4).
-
•
Decrease the colony density by plating fewer cell mass during passaging. The mean aggregate size is approximately 50–200 μm.
-
•
Avoid having the culture plate out of the incubator for more than 15 min at a time.
Problem 5
IPSCs show low attachment after plating.
Potential solution
-
•
Plate a 2–3-fold higher number of cell mass initially and maintain a more densely confluent culture.
-
•
Work quickly after cells are treated with passaging reagents (Versene) to minimize the duration that the mass are in suspension. The optimal time range is from 5–7 min.
-
•
Reduce incubation time with passaging reagents, to which some cell lines/cultures are more sensitive. This is particularly important if cells are passaged before cell multi-layering within the colony.
-
•
Do not pipette up and down more than 2–3 times when disrupting cell mass. If the desired size (50–200 μm) is not achieved by this means, increase the incubation time with the passaging reagent by 1–2 min. This is particularly important if colonies are very dense and cell mass are difficult to break up.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lidan Hu (hulidan@zju.edu.cn).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
I am extremely grateful to all members of the Mao and Hu Lab, past and present, for the interesting discussions and great contributions to the project. I thank the National Clinical Research Center for Child Health for the great support. This study was financially supported by the National Natural Science Foundation of China (U20A20351, 81770710) and the Natural Science Foundation of Zhejiang Province of China (no. LQ22C070004).
Author contributions
L.H. and J.M. conceived, designed, and supervised the research. L.G. and F.W. performed the experiments. L.G., F.W., and Y.W. recruited patients and collected samples. L.H. and L.G. performed data analyses. L.H., L.G., and J.M. wrote the manuscript. All authors have read and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Lidan Hu, Email: hulidan@zju.edu.cn.
Jianhua Mao, Email: maojh88@zju.edu.cn.
Data and code availability
The protocol including all materials and requirements during this study are available.
References
- Choi S.M., Liu H., Chaudhari P., Kim Y., Cheng L., Feng J., Sharkis S., Ye Z., Jang Y.-Y. Reprogramming of EBV-immortalized B-lymphocyte cell lines into induced pluripotent stem cells. Blood. 2011;118:1801–1805. doi: 10.1182/blood-2011-03-340620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Wang G., Wu H., Fu H., Wang Y., Yu F., Liu Z., Mao J. Establishment of an induced pluripotent stem cell line (NCKDi003-A) from a patient with X-linked Dent disease (X-Dent) carrying the hemizygote mutation p. T277P (c. 829A > C) in the CLCN5 gene. Stem Cell Res. 2021;56:102538. doi: 10.1016/j.scr.2021.102538. [DOI] [PubMed] [Google Scholar]
- Loh Y.-H., Hartung O., Li H., Guo C., Sahalie J.M., Manos P.D., Urbach A., Heffner G.C., Grskovic M., Vigneault F., et al. Reprogramming of T Cells from human peripheral blood. Cell Stem Cell. 2010;7:15–19. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack A.A., Kroboth S., Rajesh D., Wang W.B. Generation of induced pluripotent stem cells from CD34+ cells across blood drawn from multiple donors with non-integrating episomal vectors. PLoS One. 2011;6:e27956. doi: 10.1371/journal.pone.0027956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niibe K., Kawamura Y., Araki D., Morikawa S., Miura K., Suzuki S., Shimmura S., Sunabori T., Mabuchi Y., Nagai Y., et al. Purified mesenchymal stem cells are an efficient source for iPS cell induction. PLoS One. 2011;6:e17610. doi: 10.1371/journal.pone.0017610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Yamakawa T., Matsumura Y., Sato Y., Amano N., Watanabe A., Goshima N., Yamanaka S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M.A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J.L., Tian S., Nie J., Jonsdottir G.A., Ruotti V., Stewart R., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhang S.-C., Wernig M., Duncan I.D., Brüstle O., Thomson J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhou T., Benda C., Duzinger S., Huang Y., Li X., Li Y., Guo X., Cao G., Chen S., Hao L., et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011;22:1221–1228. doi: 10.1681/ASN.2011010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The protocol including all materials and requirements during this study are available.